User Manual
Overview
NMPFamsDB is a database which hosts novel metagenome protein clusters with no or weak hits to Pfam or Reference genomes and aiming at significantly expanding the protein family space known till today.
With NPFAMsDB you can:
- Explore the novel protein families and their genomic content
- See their habitat distribution
- Follow their biogeographical trademarks
- See their taxonomy information
- Explore potential novel structures and folds
- Use Hidden Markov Model (HMM) and sequence-based searches for querying the protein clusters
- Filter information by smart filters at any stage of analysis
NMPFamsDB has the following pages/sections:
- Home: The NMPFamsDB Home Page
- Browse: The database browser. You can browse NMPFamsDB contents from the following subcategories:
- Sequence Search Tools: Perform sequence-based queries. The following options are offered:
- LAST Search: Perform pairwise alignment searches using LAST.
- HMMER Search: Perform HMM-powerred sequence searches using HMMER.
- Pattern Search: search sequences using PROSITE-like sequence patterns or regular expressions.
- Visualization Tools: Perform various types of analyses. The following options are offered:
- Ecosystem & Phylogeny: Create various types of plots for subsets of the database.
- Geographical Distribution: Visualize the geographical distribution of database components.
- Programmatic Access: The NMPFamsDB Application Programming Interface (API).
- Statistics: An overview of NMPFamsDB's contents and their distributions.
- Downloads: Download NMPFamsDB contents in various file formats.
- Manual: This Help Section.
- Contact: The database's contact information.
Availability
NMPFamsDB is publicly available through http://nmpfamsdb.pavlopouloslab.info or https://bib.fleming.gr/NMPFamsDB.
How to use this manual
Topics in the Manual are divided in separate tabs, accessible through header buttons at the top of the page. Click on any of these header buttons to navigate to its respective section.
Long sections are divided into subsections, that can be scrolled up and down. At the end of each subsection a link exists, labeled [Back to top]. Clicking on it will return you to the top of the section.
Data organization in NMPFamsDB
The data contained in NMPFamsDB falls under the following four categories: Families, Sequences, Datasets and Habitats.
-
Novel Metagenome Protein families (or NMPFs) are the main entry category of NMPFamsDB. They represent collections of protein sequences, clustered based on their sequence identity. Each NMPF is characterized by a consensus sequence, a number of Multiple Sequence Alignments (MSAs) and an associated Hidden Markov Model (HMM) profile, secondary structure and protein topology predictions, phylogenetic, ecosystem and geographical distribution annotations. In addition, a subset of NMPFs have available 3D structure models predicted using AlphaFold. NMPFs are represented by unique alphanumerical identifiers (e.g. F000001, F000018, F134500) or by their corresponding numerical indexes (e.g. 1, 18, 134500). A detailed description on the information contained for each family is given in the section “Protein Families”.
-
Protein sequences are given in NMPFamsDB both as family members and as individual entities. Each sequence is characterized by a number of identifiers, namely, a unique Protein ID, corresponding to the IMG JGI Gene OID, a Dataset Taxon OID, corresponding to the JGI Taxon OID of environmental samples, and a Scaffold ID, corresponding to the equivalent JGI Scaffold OID for sequencing contig/scaffolds. A detailed description on sequences is given in the section “Sequences”.
-
Scaffolds represent the sequencing contigs derived from a metagenome or metatranscriptome dataset. from which the protein sequences were derived. Each Scaffold is represented by a Taxon OID, corresponding to the equivalent IMG JGI Taxon OID for sequencing datasets. A detailed description on the information contained for each scaffold is given in the section “Scaffolds”.
-
Datasets represent the Metagenome (or Metatranscriptome) sequencing samples/datasets from which the associated scaffolds and protein sequences were derived. Each Dataset is represented by a Taxon OID, corresponding to the equivalent IMG JGI Taxon OID for sequencing datasets. Additional information includes ecosystem classification, geographical metadata and availability for public use. A detailed description on the information contained for each dataset is given in the section “Datasets”.
-
Habitats represent the documented ecosystems of the sequenced datasets from which protein sequences have been collected. They are hierarchically organized, grouped into categories based on their nature and characteristics. Habitats are directly linked to the metagenome datasets and, through them, to scaffolds, protein sequences and NMPFs. A detailed description on sequences is given in the section “Ecosystems”.
Browsing and Searching NMPFamsDB
Users can browse NMPFamsDB’s contents through the web interface’s “Browse” sub-menu in fivw different manners, by Family, Sequence, Scaffold, Dataset and Habitat:
-
Browse Families: The database’s novel metagenome protein families (NMPFs).
-
Browse Sequences: All sequences in the database.
-
Browse Sequences: The sequencing scaffolds from which sequence data has been collected.
-
Browse Datasets: The metagenome and metatrasncriptome datasets from which sequence data has been collected.
-
Browse Habitats: The ecosystems from which datasets were obtained and sequences were derived.
Families
Browse Families
By choosing Browse → Families you will be redirected to the following page:

The top of the page contains a search form with a number of options that can help you filter data according to your needs. Browse / search results are presented in a table containing the following information:
- Numeric index: The entry’s numeric index in the search
- ID: The entry’s Family ID. The ID is clickable, leading to the respective family's entry page.
- Dataset Category: The family’s category, based on the dataset type(s) from where sequences were collected. Available types are: Metagenome, Metatranscriptome and Metagenome / Metatranscriptome.
- Number of Sequences: The number of sequences contained in the family
- Number of Datasets: The number of associated datasets.
- Number of Scaffolds: The number of associated scaffolds.
- 3D Model: An indicator on whether the family has an available 3D structure ('Y') or not ('N').
Users can view a family’s entry page by clicking on its ID. The family’s Number of Sequences, Datasets and Scaffolds are also clickable; by selecting them users can view all sequences, datasets or scaffolds associated with a particular family.
Browse and search results are organized in pages. Users can select the number of entries per page by selecting a value in the “Show Entries” drop-down menu, located at the top left side of the entries table.
One or multiple entries can be selected by clicking their checkboxes, appearing at the start of each line. By clicking the checkbox in the table's head line, all entries per page can be selected at once. Selected entries can then be downloaded, using the options presented at the top right side of the table. Data can be downloaded in either tab (TSV) or comma-delimited (CSV) format, by choosing the appropriate option and clicking the “Export” button.
Family searches can be performed by using the search form in the top of the page. The form contains four panels, corresponding to four search capabilities, “Keyword”, "Sequence & Structure", “Environment” and "Phylogeny".
Keyword
The following options are offered in the Keyword section of the search form:
- Family ID(s): Search families based on their identifiers. Input one or more family IDs separated by spaces. Both full IDs (e.g.
F000015) and family number IDs (e.g.15) can be used. The search will return the families of the submitted IDs. - IMG/M Taxon OID(s): Search families based on their associated IMG/M datasets. Input one or more Taxon OIDs (e.g.
3300004768) separated by spaces. The search will return all families that are contain sequences from the queried datasets. - IMG/M Scaffold ID(s): Search families based on their associated IMG/M scaffolds. Input one or more Scaffold IDs (e.g.
JGI20166J26741_10000139) separated by spaces. The search will return all families that are contain sequences from the queried scaffolds. - IMG/M Gene/Protein ID(s): Search families based on their sequence contents. Input one or more protein IDs (e.g.
GI24668J20090_102382961) separated by spaces. The search will return the families containing the submitted sequences. - Family Category: Search families based on their sequences' sources of origin. Available categories are:
- All: return all families regardless of their category
- Metagenome: return families containing only sequences from metagenomes
- Metatranscriptome: return families containing only sequences from metatranscriptomes
- Metagenome / Metatranscriptome: return "mixed" families containing sequences from both metagenomes and metatranscriptomes.
- Number of datasets: Search families based on the number of associated datasets. Click the checkbox to activate the slider and then drag its two endpoints to define the minimum and maximum value. Default values are
1(minimum) and1195maximum.
Sequence & Structure
- Number of sequences: Search families based on the number of sequence members. Click the checkbox to activate the slider and then drag its two endpoints to define the minimum and maximum value. Default values are
100(minimum) and16856maximum. - Sequence length: Search families based on their average sequence length. Click the checkbox to activate the slider and then drag its two endpoints to define the minimum and maximum value.
- Predicted Topology: Search families based on their predicted protein topology. Check all fields that apply to limit the search to specific topology types. Leaving everything checked or unchecked will search all categories. Available topology types include:
- Globular: Soluble globular proteins.
- Transmembrane: Proteins with transmembrane segments spanning a lipid bilayer.
- Fibrus: Fibrillar proteins.
- Mixed: Proteins with mixed features, containing both globular domains and fibrillar segments.
- Predicted 3D structure: Search families based on whether an AlphaFold 3D structure model is available or not. Check 'Yes' for limiting the search to families with 3D models or 'No' to search for families without models. Leaving both options checked or unchecked will search all categories.
- Structure confidence: Limit the search of families with 3D models (see previous options) based on their total structure confidence score (pLDDT), as calculated by AlphaFold. pLDDT ranges from 0 to 100, with higher values indicating stronger confidence. Click the checkbox to activate the slider and then drag its two endpoints to define the minimum and maximum value.
Environment
The Environment panel enables you to perform searches based on the families' associations with ecosystems, based on the analyzed metagenome and metatranscriptome datasets. All habitats are shown in a hierarchical tree structure, following the GOLD ecosystem classification system. The available ecosystems belong to one of the following general categories:
- Environmental: contains the ecosystems of datasets coming from environmental samples,e.g. water, soil etc.
- Host-associated: contains ecosystems of datasets obtained from organisms acting as hosts to microbes, e.g. humans, mammals, plants etc.
- Engineered: contains a number of environments that were man-made, e.g. factories, industrial wastes, lab wastes etc.
Each category is further divided into sub-categories that can be displayed or hidden by clicking on the red caret ( ) icons. You can also expand or collapse the entire tree by clicking on the Expand All or Collapse All button, respectively.
To select an ecosystem for search, simply click the checkbox next to its name. You can select multiple ecosystems to perform complex queries. The names of the selected ecosystems will appear in the list box at the right of the tree.
Most families are not limited to a single ecosystem; in fact, they are associated with multiple ecosystem types, with each type appearing at a particular association level (for example, a family can be 30% Environmental and 70% Host-associated). You can further limit your query options and search families that are associated with an ecosystem above a certain percentage cut-off, simply by clicking the "Association cut-off" checkbox and selecting a cut-off level by dragging the slider button. A small subset of the families are associated with a single ecosystem type (e.g. 100% association with plants). You can select to retrieve only these families by activating the checkbox marked "Limit search to families containing sequences from only the selected environments". This is the equivalent to setting the Association cut-off slider at 100%.
Phylogeny
Similar to the ecosystem search, you can perform searches based on phylogenetic associations. All organisms associated with sequences in NMPFamsDB are shown in a hierarchical tree structure, following the NCBI nomenclature, classification system and ranking. Like in Ecosystem search, you can expand or collapse a category by clicking on the red caret ( ) icon or by using the Expand All or Collapse All buttons. You can select one or more taxa by clicking the checkbox next to their names. The selected taxa will appear in the list box at the right of the tree.
Performing a search query
To perform a search query, simply set the values you wish to search in each of the respective fields. The query options of the Keyword, Sequence & Structure, Environment and Phylogeny panels can be combined. For example, you can choose to search for families coming only from Metagenomes that have an available 3D structure, are associated with Host-associated ecosystems, and contain sequences from viruses, by setting the appropriate options, as in the figure below:

When you are ready to begin, click the "Submit" button at the bottom of the form. To click all options, click on the "Reset" button. After the search is completed, a table of results will appear, in the same format as in the original page. In addition, the query options will appear in a box marked "Search Options".
Family Entry Organization
Each family entry is divided in distinct sections. You can access them by navigating the family page or by using the buttons at the top of the entry. At the top right of the screen you will find the family's downloadable files. You can download a file simply by choosing it from the selection list and clicking the "Download" button.
Overview
The first section presents an overview of the family's properties. These include the family identifier and category, the number of associated sequences, datasets and scaffolds, the average sequence length and the representative consensus sequence, derived from the family's multiple sequence alignment.
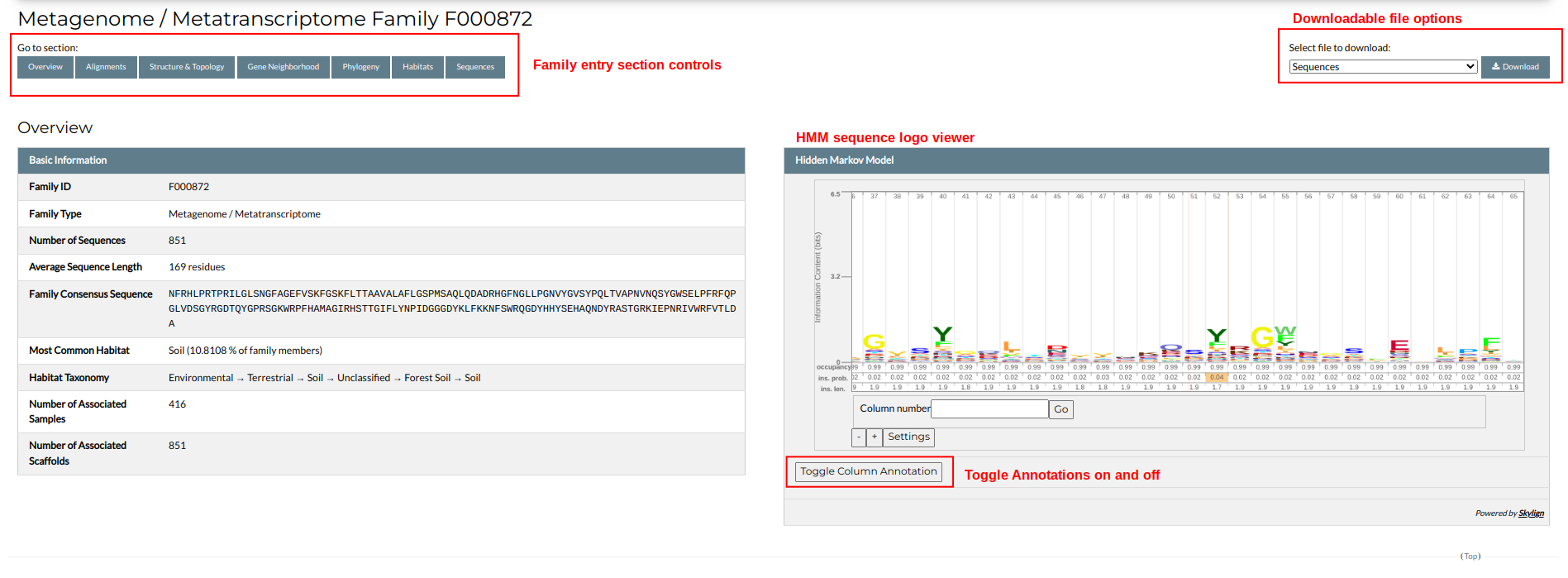
The right side of the section contains an interactive viewer displaying the family's Hidden Markov Model (HMM) profile in a Sequence Logo representation. In the logo, each column corresponds to an alignment / HMM position and contains the residues found in that position, colored based on their type. Each amino acid's size in a column is proportional to its probability to be found in that particular position. You can scroll through the HMM viewer by left-clicking and dragging the Sequence Logo. Clicking on a position will open a window showing a detailed list of all residue probabilities. These can also be toggled on and off by clicking the "Toggle Column Annotation" button.
Alignments
This section contains an interactive viewer for the family's Multiple Sequence Alignments (MSAs). In NMPFamsDB each family is represented by two alignments; a full MSA, containing all the family's sequences and a "seed" MSA, containing a representative, non-redundant subset of the sequences. The seed MSA is also the alignment used to create the family's HMM profile. You can access each MSA by clicking on the relevant button a the top of the viewer.
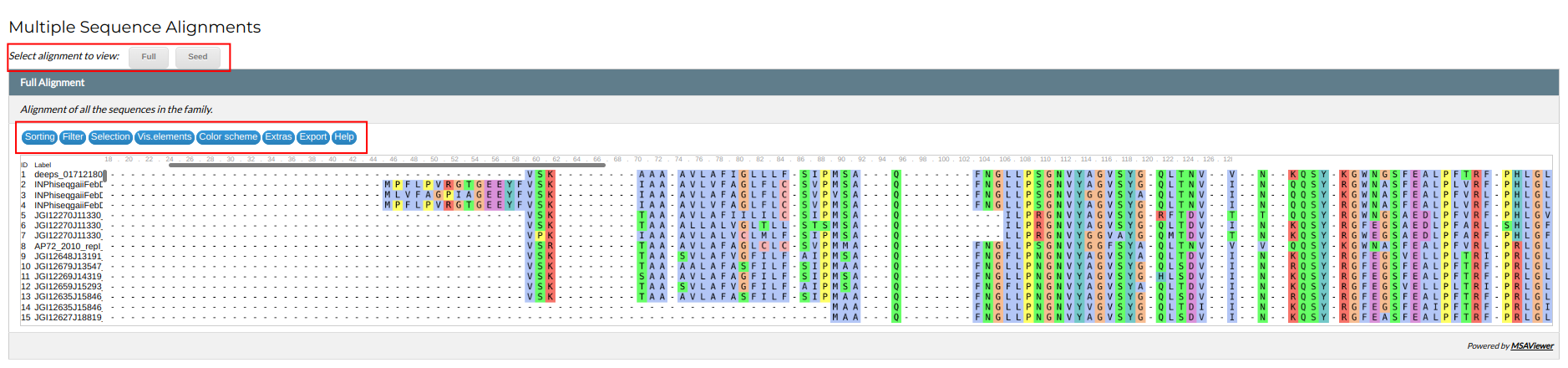
The MSA in the viewer can be scrolled by using the mouse. At the top of the viewer, a menu presents a number of functions to visualize and manipulate the alignment:
- Sorting: sort the sequences in the alignment based ID or name.
- Filter: filter the alignment components by sequence identity, gaps, or custom search queries.
- Vis. elements: show or hide alignment elements .
- Color scheme: choose a coloring scheme for the alignment.
- Extras: add the consensus sequence in the alignment or jump to a specific position.
- Export: export the alignment or a subset of the alignment in various file formats.
- Help: access the alignment viewer help page.
Structure & Topology
The structure and topology section presents the structural annotation of the family.
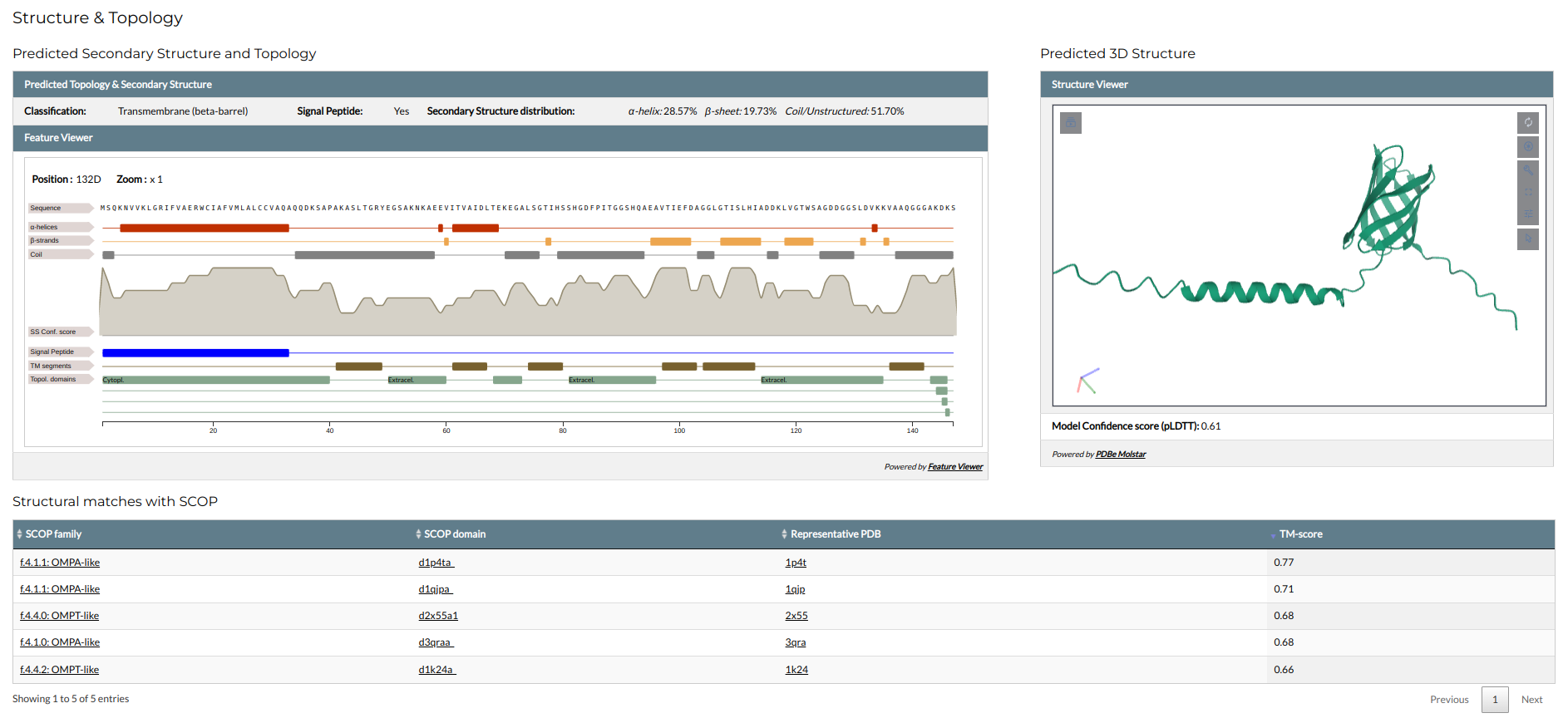
- all families have annotations on their secondary structure and topology, predicted through a number of prediction methods based on their amino acid sequence. These are shown in an interactive feature viewer and include the following:
- Classification: Predicted family topology. Families can be globular, transmembrane, fibrous or mixed globular fibrous.
- if the family is predicted as transmembrane, its category is also given in parentheses. Transmembrane proteins can be α-helical (predicted by Phobius) or β-barrels (predicted by PRED-TMBB2). The family's transmembrane topology is shown in the viewer.
- Signal Peptide: Indication on whether the family sequences contain a signal peptide predicted by SignalP. If a signal peptide is predicted, its position is shown in the viewer.
- Secondary structure: the secondary structure elements of the family, as predicted through Porter5. Although a number of different secondary structure components exist (α-helices, 3~10~-helices, β-strands, β-bridges, turns, bends etc.), for efficiency they are grouped into three categories (α-helices, β-strands forming β-sheets and coil/unstructured). The secondary structure topology is shown in the viewer, accompanied by a graph displaying its confidence score.
- In cases where sequences have intrinsically disordered regions (predicted with mobidb-lite), they are also shown in the viewer as a distinct field.
- Classification: Predicted family topology. Families can be globular, transmembrane, fibrous or mixed globular fibrous.
- If the family has a 3D structure predicted with AlphaFold, it is shown through an interactive structure viewer (Molstar) at the right. The confidence of the structure (pLDDT), as calculated by AlphaFold, is also given. The viewer can be controlled using the mouse.
- Families with 3D models and high AlphaFold confidence (pLDDT >=0.7) have been subjected to structural alignments against the SCOP family of domains. These alignments were performed with TM-align (or MM-align for multi-domain structures) and the TM-score of the alignment was used to define structure similarity. If a structure has hits to SCOP (TM-score >=0.50), they are presented in a table below the feature and structure viewers. The table shows the IDs and names of the matching SCOP superfamily and domains, as well as the PDB ID of a representative structure in the Protein Data Bank. The TM-score of each match is also given.
Gene Neighborhood

If a family contains sequences from scaffolds that also have hits to Pfam domains, these domains are considered to be the family's gene neighborhood. The Pfam IDs and names of these domains, as well as their % frequency in the family's associated scaffolds, are given in a table.
Phylogeny
This section presents the phylogenetic annotation of the family, based on the metadata of the associated scaffolds.
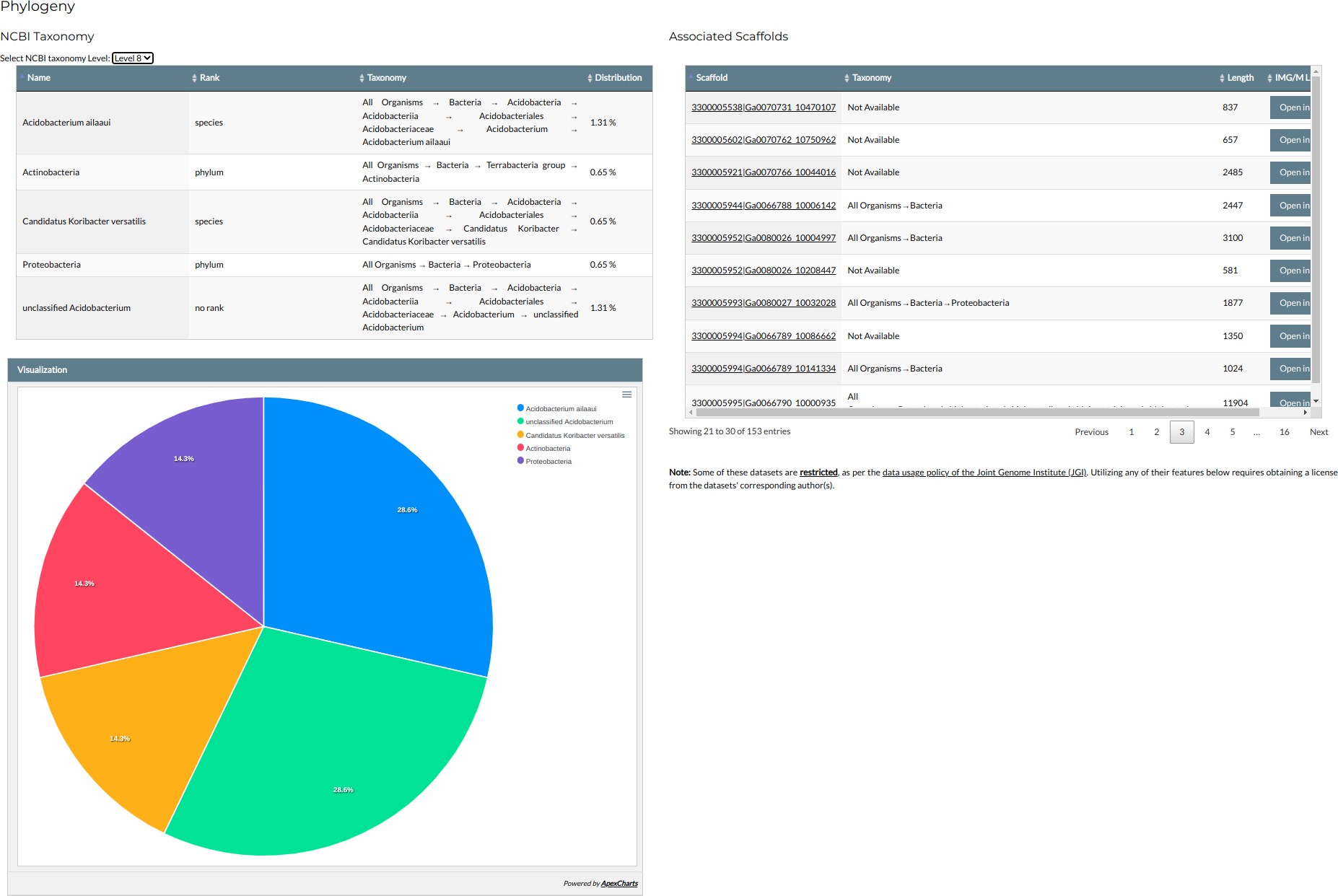
- The % distribution of taxa in the family is shown in an interactive table and associated pie chart at the left of the section. The taxonomic level, following the NCBI ranking, can be chosen through the selection list at the top of the table.
- The family's associated scaffolds and their taxonomy are shown at the right of the section. Clicking on the scaffolds ID will redirect you to each scaffold's page in NMPFamsDB (see the "Scaffolds" section for details). Clicking on the "Open in IMG/M" button will open a window to the scaffold's page in IMG/M.
Habitats
This section presents the ecosystem annotation of the family, based on the metadata of the associated datasets. It is further divided into two parts, Associated Habitat Types and Associated Datasets.
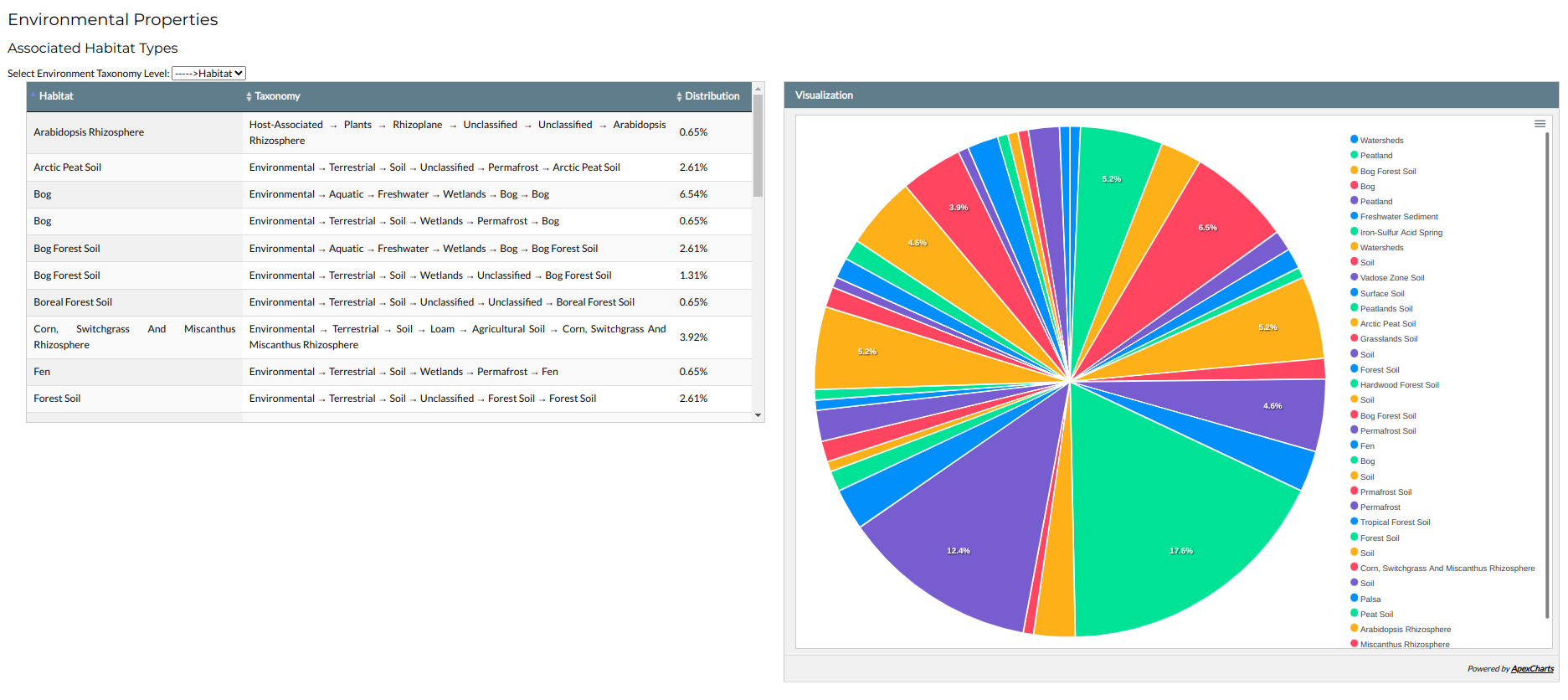
The first part of the section presents the % distribution of ecosystems in the family, through an interactive table and an associated pie chart. Like the Phylogeny section, the various levels of habitat taxonomy can be accessed through the selection list a the top of the table.
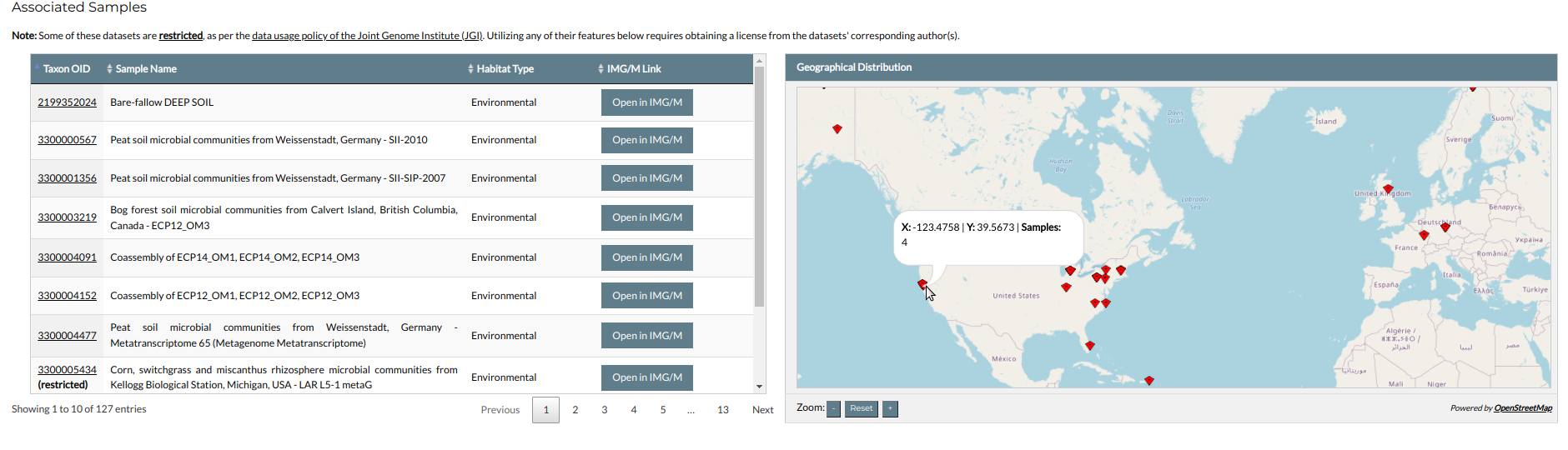
The second part shows the family's associated datasets in a table, accompanied by an interactive map showing their geographical distribution. Clicking on a dataset's Taxon OID will redirect you to its page in NMPFamsDB (see the "Datasets" section for details). In the map, dataset locations are indicated by red markers. Hovering the mouse over a marker will open a pop-up window showing its coordinates, as well as the number of sampled datasets from that location.
Sequences
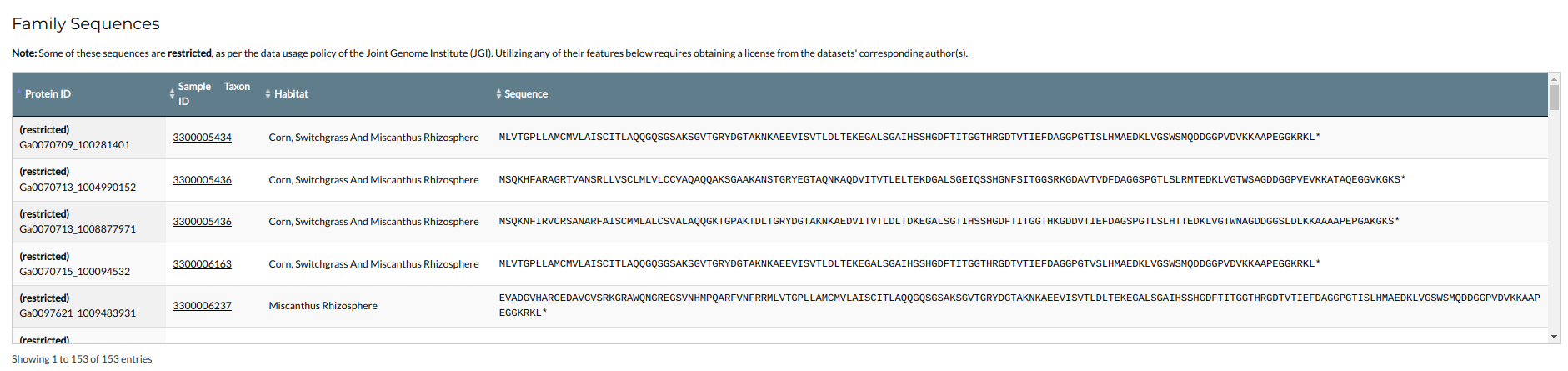
The final section shows the family's protein components. Each protein is represented by its identifiers (Protein ID, Taxon OID and Scaffold ID), its environmental metadata and its amino acid sequence.
Downloadable Files
Each family has a number of files available for download through the form at the top right corner of the page. These include:
- The family's protein sequences in FASTA format.
- The family's full and seed MSAs in FASTA format.
- The family's HMM profile in the HMMER or HH-suite formats.
- The family's 3D model (if available) in the PDB format.
All files are encoded in ASCII UTF-8 and can be opened by text editors, or manipulated using specialized software:
- sequence and MSA files can be opened by sequence or alignment viewers (e.g. Jalview, ClustalX, BioEdit etc.)
- The HMMER HMM profiles are compatible with the HMMER 3 suite and can be used with tools such as hmmscan, hmmsearch or hmmalign.
- The HH-suite HMM profiles can be used with HH-suite tools such as hhblits or hhsearch.
- The PDB models can be visualized with 3D molecular viewers such as PyMOL, UCSF Chimera or VMD.
Sequences
Browse sequences
By choosing Browse → Sequences you will be redirected to the following page:

The top of the page contains a search form with a number of options that can help you filter data according to your needs. Browse / search results are presented in a table containing the following information:
- Numeric index: The sequence's numeric index in the search
- Protein ID: The sequence's Protein ID. This is by default the Gene ID of the sequence in the IMG/M database.
- Family: The ID of the family containing this sequence. The Family ID is clickable; clicking it will redirect you to the respective Family entry.
- Taxon OID: The Taxon OID of the metagenome/metatranscriptome dataset from which this sequence was derived. The ID is clickable; clicking it will redirect you to the associated Dataset entry.
- Scaffold ID: The ID of the sequencing scaffold containing the gene of this sequence. The ID is clickable; clicking it will redirect you to the associated Scaffold entry.
- Length: The sequence's length (number of residues)
- Habitat: Environmental annotation of the sequence.
- Phylogeny: Taxonomic association of the sequence. If no taxonomy is known, this section is labeled "Unclassified".
You can see each sequence by clicking the Show/Hide button to expand and collapse it in each table row.
Browse and search results are organized in pages. Users can select the number of entries per page by selecting a value in the “Show Entries” drop-down menu, located at the top left side of the entries table.
One or multiple entries can be selected by clicking their checkboxes, appearing at the start of each line. By clicking the checkbox in the table's head line, all entries per page can be selected at once. Selected entries can then be downloaded, using the options presented at the top right side of the table. Data can be downloaded in tab (TSV) or comma-delimited (CSV) format, as well as in the FASTA sequence format, by choosing the appropriate option and clicking the “Export” button.
Like families, sequence searches can be performed by using the search form in the top of the page. The form contains three panels, corresponding to three search capabilities, “Keyword”, “Environment” and "Phylogeny".
Keyword
The following options are offered in the Keyword section of the search form:
- IMG/M Gene/Protein ID(s): Retrieve sequences based on their Protein IDs. Input one or more protein IDs (e.g.
GI24668J20090_102382961) separated by spaces. The search will return the sequences of the submitted IDs. - Family ID(s): Search sequences based on their family classification. Input one or more family IDs separated by spaces. Both full IDs (e.g.
F000015) and family number IDs (e.g.15) can be used. The search will return the sequences belonging to the submitted IDs. - IMG/M Taxon OID(s): Search sequences based on their associated IMG/M datasets. Input one or more Taxon OIDs (e.g.
3300004768) separated by spaces. The search will return the sequences belonging to the submitted datasets. - IMG/M Scaffold ID(s): Search sequences based on their associated IMG/M scaffolds. Input one or more Scaffold IDs (e.g.
JGI20166J26741_10000139) separated by spaces. The search will return the sequences belonging to the submitted scaffolds. - Sample Type: Search sequences based on their source dataset type. Available categories are:
- Metagenome: return sequences coming from metagenomes
- Metatranscriptome: return sequences coming from metatranscriptomes
-
- Sequence length: Search sequences based on their length. Drag the slider's two endpoints to define the minimum and maximum value.
Environment
The Environment panel enables you to perform searches using the sequences' ecosystem annotation, based on the analyzed metagenome and metatranscriptome datasets. All habitats are shown in a hierarchical tree structure, following the GOLD ecosystem classification system. The available ecosystems belong to one of the following general categories:
- Environmental: contains the ecosystems of datasets coming from environmental samples,e.g. water, soil etc.
- Host-associated: contains ecosystems of datasets obtained from organisms acting as hosts to microbes, e.g. humans, mammals, plants etc.
- Engineered: contains a number of environments that were man-made, e.g. factories, industrial wastes, lab wastes etc.
Each category is further divided into sub-categories that can be displayed or hidden by clicking on the red caret ( ) icons. You can also expand or collapse the entire tree by clicking on the Expand All or Collapse All button, respectively.
To select an ecosystem for search, simply click the checkbox next to its name. You can select multiple ecosystems to perform complex queries. The names of the selected ecosystems will appear in the list box at the right of the tree.
Phylogeny
Similar to the ecosystem search, you can perform searches based on phylogenetic associations. All organisms associated with sequences in NMPFamsDB are shown in a hierarchical tree structure, following the NCBI nomenclature, classification system and ranking. Like in Ecosystem search, you can expand or collapse a category by clicking on the red caret ( ) icon or by using the Expand All or Collapse All buttons. You can select one or more taxa by clicking the checkbox next to their names. The selected taxa will appear in the list box at the right of the tree.
Performing a search query
To perform a search query, simply set the values you wish to search in each of the respective fields. The query options of the Keyword, Environment and Phylogeny panels can be combined. For example, you can choose to search for Metagenome sequences that are associated with Engineered ecosystems, and contain sequences from Archaea, by setting the appropriate options, as in the figure below:

Scaffolds
Browse Scaffolds
By choosing Browse → Scaffolds you will be redirected to the following page:

The top of the page contains a search form with a number of options that can help you filter data according to your needs. Browse / search results are presented in a table containing the following information:
- Numeric index: The entry's numeric index in the search
- Scaffold: The scaffold's name, typically shown in the format "Taxon OID|Scaffold ID". The name is clickable; clicking it will redirect you to the respective Scaffold entry.
- Family: The ID of the family containing this sequence. The Family ID is
- Taxon OID: The Taxon OID of the metagenome/metatranscriptome dataset from which this scaffold was derived. The ID is clickable; clicking it will redirect you to the associated Dataset entry.
- Scaffold ID: The Scaffold ID.
- Scaffold Length: The scaffold length in base pairs (bps)
- Associated Families: the number of associated families.
- Associated Sequences: the number of associated sequences.
- NCBI Taxonomy: Taxonomic association of the sequence. If no taxonomy is known, this section is labeled "Unclassified".
Browse and search results are organized in pages. Users can select the number of entries per page by selecting a value in the “Show Entries” drop-down menu, located at the top left side of the entries table.
One or multiple entries can be selected by clicking their checkboxes, appearing at the start of each line. By clicking the checkbox in the table's head line, all entries per page can be selected at once. Selected entries can then be downloaded, using the options presented at the top right side of the table. Data can be downloaded in tab (TSV) or comma-delimited (CSV) format, by choosing the appropriate option and clicking the “Export” button.
Scaffold searches can be performed by using the search form in the top of the page. The form contains three panels, corresponding to three search capabilities, “Keyword”, “Environment” and "Phylogeny".
Keyword
The following options are offered in the Keyword section of the search form:
-
IMG/M Taxon OID(s): Search based on associated IMG/M datasets. Input one or more Taxon OIDs (e.g.
3300004768) separated by spaces. The search will return the scaffolds belonging to the submitted datasets. -
IMG/M Scaffold ID(s): Retrieve scaffolds based on their Scaffold IDs. Input one or more Scaffold IDs (e.g.
JGI20166J26741_10000139) separated by spaces. The search will return the scaffolds of the specified IDs. -
IMG/M Gene/Protein ID(s): Search based on sequence components. Input one or more protein IDs (e.g.
GI24668J20090_102382961) separated by spaces. The search will return the scaffolds containing of the submitted IDs. -
Family ID(s): Search scaffolds based on their associated. Input one or more family IDs separated by spaces. Both full IDs (e.g.
F000015) and family number IDs (e.g.15) can be used. The search will return the scaffolds associated with the submitted IDs. -
Family Category: Search sequences based on their associated dataset type. Available categories are:
- Metagenome: return sequences coming from metagenomes
- Metatranscriptome: return sequences coming from metatranscriptomes
-
Scaffold length: Search scaffolds based on their length. Drag the slider's two endpoints to define the minimum and maximum value.
Environment
The Environment panel enables you to perform searches using the scaffolds' ecosystem annotation, based on the analyzed metagenome and metatranscriptome datasets. All habitats are shown in a hierarchical tree structure, following the GOLD ecosystem classification system. The available ecosystems belong to one of the following general categories:
- Environmental: contains the ecosystems of datasets coming from environmental samples,e.g. water, soil etc.
- Host-associated: contains ecosystems of datasets obtained from organisms acting as hosts to microbes, e.g. humans, mammals, plants etc.
- Engineered: contains a number of environments that were man-made, e.g. factories, industrial wastes, lab wastes etc.
Each category is further divided into sub-categories that can be displayed or hidden by clicking on the red caret ( ) icons. You can also expand or collapse the entire tree by clicking on the Expand All or Collapse All button, respectively.
To select an ecosystem for search, simply click the checkbox next to its name. You can select multiple ecosystems to perform complex queries. The names of the selected ecosystems will appear in the list box at the right of the tree.
Phylogeny
Similar to the ecosystem search, you can perform searches based on phylogenetic associations. All organisms associated with scaffolds in NMPFamsDB are shown in a hierarchical tree structure, following the NCBI nomenclature, classification system and ranking. Like in Ecosystem search, you can expand or collapse a category by clicking on the red caret ( ) icon or by using the Expand All or Collapse All buttons. You can select one or more taxa by clicking the checkbox next to their names. The selected taxa will appear in the list box at the right of the tree.
Performing a search query
To perform a search query, simply set the values you wish to search in each of the respective fields. The query options of the Keyword, Environment and Phylogeny panels can be combined. For example, you can choose to search for Metagenome scaffolds that are associated with Engineered ecosystems, and contain sequences from Archaea, by setting the appropriate options, as in the figure below:
Scaffold Entry Organization
The Scaffold entry page contains the basic information on the sequencing scaffold and is divided into four sections: Overview, Ecosystem & Geography, Associated Families and Sequences.
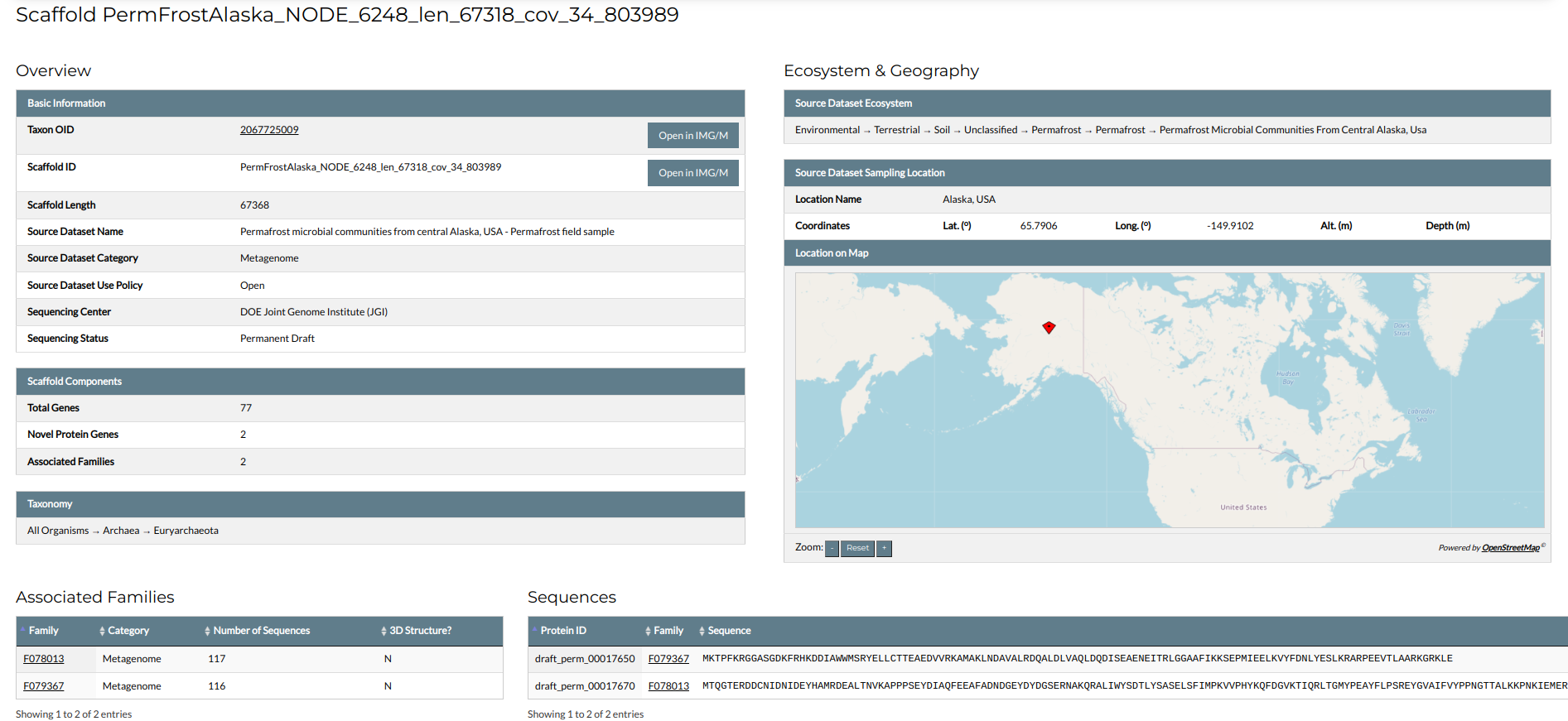
Overview
The Overview section contains the basic properties of the scaffold. These include the scaffolds Taxon OID and Scaffold ID, and its length in base pairs (bps). Links to the associated dataset and scaffold pages in IMG/M are offered through the "Open in IMG/M" buttons. In addition, the basic metadata of the source metagenomic dataset are presented, namely, the name of the dataset, its category (Metagenome or Metatranscriptome), its data usage policy, and information on the sequencing procedure. Apart from the above, the section gives an overview on the components of the scaffold: the total number of genes, the number of novel protein genes, and then number of associated NMPFs. If the taxonomy of the scaffold is known, it is given in the box titled "Taxonomy"; otherwise, it is characterized as "Unclassified".
Ecosystem & Geography
The Ecosystem & Geography section presents the ecosystem metadata of the scaffold, as defined from the source dataset. The scaffold's ecosystem habitat is shown in the GOLD classification system. In addition, the geographic location of the sample is presented, both in the form of coordinates (longitude, latitude and if applicable, altitude or depth), and in the form of an interactive map panel.
Associated Families
The Associated Families section gives a list of all families containing at least one of the scaffold's novel protein genes.
Sequences
This section gives a list with all the scaffold's novel protein sequences.
Datasets
Browse Datasets
By choosing Browse → Datasets you will be redirected to the following page:

The top of the page contains a search form with a number of options that can help you filter data according to your needs. Browse / search results are presented in a table containing the following information:
- Numeric index: The entry's numeric index in the search
- Taxon OID: The Taxon OID of the metagenome/metatranscriptome dataset . The name is clickable; clicking it will redirect you to the respective dataset entry.
- Sample Name: The name of the metagenomic dataset.
- Sample Type: The type of the dataset (Metagenome or Metatranscriptome).
- Associated Families: the number of associated families.
- Associated Sequences: the number of associated sequences.
- Habitat: The ecosystem from which the dataset was collected.
- Location: The dataset's sampling location.
Browse and search results are organized in pages. Users can select the number of entries per page by selecting a value in the “Show Entries” drop-down menu, located at the top left side of the entries table.
One or multiple entries can be selected by clicking their checkboxes, appearing at the start of each line. By clicking the checkbox in the table's head line, all entries per page can be selected at once. Selected entries can then be downloaded, using the options presented at the top right side of the table. Data can be downloaded in tab (TSV) or comma-delimited (CSV) format, by choosing the appropriate option and clicking the “Export” button.
Searches can be performed by using the search form in the top of the page. The form contains three panels, corresponding to three search capabilities, “Keyword”, “Environment” and "Phylogeny".
Keyword
The following options are offered in the Keyword section of the search form:
- IMG/M Taxon OID(s): Retrieve datasets based on their IMG/M datasets. Input one or more Taxon OIDs (e.g.
3300004768) separated by spaces. The search will return the datasets corresponding to the Taxon OIDs. - IMG/M Scaffold ID(s): Retrieve datasets based on their associated scaffolds. Input one or more Scaffold IDs (e.g.
JGI20166J26741_10000139) separated by spaces. - IMG/M Gene/Protein ID(s): Search based on sequence components. Input one or more protein IDs (e.g.
GI24668J20090_102382961) separated by spaces. - Family ID(s): Search datasets based on their associated. Input one or more family IDs separated by spaces. Both full IDs (e.g.
F000015) and family number IDs (e.g.15) can be used. The search will return the datasets with sequences contained in the submitted families. - IMG/M Sample Name: Search based on the sample's name. This is a free text search field. Enter a term (e.g.
Lake) to return all datasets containing it in their name. - Sample Type: Search based on the dataset type (Metagenome or Metatranscriptome)
- Sequencing Center: search datasets based on the sequencing center. Choose a sequencing center from the list.
- Location: Search datasets based on their location. Select a sampling location from the list or enter a search term in the accompanying text field (e.g.
Canada). - Show only published samples: Some datasets in NMPFamsDB are associated to publications, while others are in draft form (temporary or permanent). To show only published datasets, check this option.
Environment
The Environment panel enables you to perform searches using the scaffolds' ecosystem annotation, based on the analyzed metagenome and metatranscriptome datasets. All habitats are shown in a hierarchical tree structure, following the GOLD ecosystem classification system. The available ecosystems belong to one of the following general categories:
- Environmental: contains the ecosystems of datasets coming from environmental samples,e.g. water, soil etc.
- Host-associated: contains ecosystems of datasets obtained from organisms acting as hosts to microbes, e.g. humans, mammals, plants etc.
- Engineered: contains a number of environments that were man-made, e.g. factories, industrial wastes, lab wastes etc.
Each category is further divided into sub-categories that can be displayed or hidden by clicking on the red caret ( ) icons. You can also expand or collapse the entire tree by clicking on the Expand All or Collapse All button, respectively.
To select an ecosystem for search, simply click the checkbox next to its name. You can select multiple ecosystems to perform complex queries. The names of the selected ecosystems will appear in the list box at the right of the tree.
Phylogeny
Similar to the ecosystem search, you can perform searches based on phylogenetic associations. All organisms associated with scaffolds in NMPFamsDB are shown in a hierarchical tree structure, following the NCBI nomenclature, classification system and ranking. Like in Ecosystem search, you can expand or collapse a category by clicking on the red caret ( ) icon or by using the Expand All or Collapse All buttons. You can select one or more taxa by clicking the checkbox next to their names. The selected taxa will appear in the list box at the right of the tree.
Performing a search query
To perform a search query, simply set the values you wish to search in each of the respective fields. The query options of the Keyword, Environment and Phylogeny panels can be combined. For example, you can choose to search for Metagenome datasets that are associated with Engineered ecosystems, and contain sequences from Archaea, by setting the appropriate options, as in the figure below:
Dataset Entry Organization
The Dataset entry page contains the basic information on the dataset sample and is divided into five sections: Overview, Ecosystem & Geography, Associated Families, Associated Scaffolds and Sequences.

Overview
The Overview section contains the basic properties of the dataset. These include the Taxon OID and metadata such as the sample name, its sequencing status and its use licence. Links to the associated dataset and scaffold pages in IMG/M are offered through the "Open in IMG/M" buttons. Apart from the above, the section gives an overview on the components of the dataset: its category (metagenome or metatranscriptome), the total metagenome size, the number of associated scaffolds, the number of novel protein genes, and then number of associated NMPFs. A metagenome sample contains genomic material from multiple organisms found in the same sample. The phylogenetic evidence for each dataset component, based on the associated scaffolds, is given in the table "Dataset Phylogeny".
Ecosystem & Geography
The Ecosystem & Geography section presents the ecosystem metadata of the scaffold, as defined from the source dataset. The scaffold's ecosystem habitat is shown in the GOLD classification system. In addition, the geographic location of the sample is presented, both in the form of coordinates (longitude, latitude and if applicable, altitude or depth), and in the form of an interactive map panel.
Associated Families
The Associated Families section gives a list of all families containing at least one of the dataset's novel protein genes.
Associated Scaffolds
The Associated Families section gives a list of all sequencing scaffolds derived from the dataset.
Sequences
This section gives a list with all the scaffold's novel protein sequences.
Ecosystems
Browse Ecosystems
By choosing Browse → Ecosystems you will be redirected to the following page:
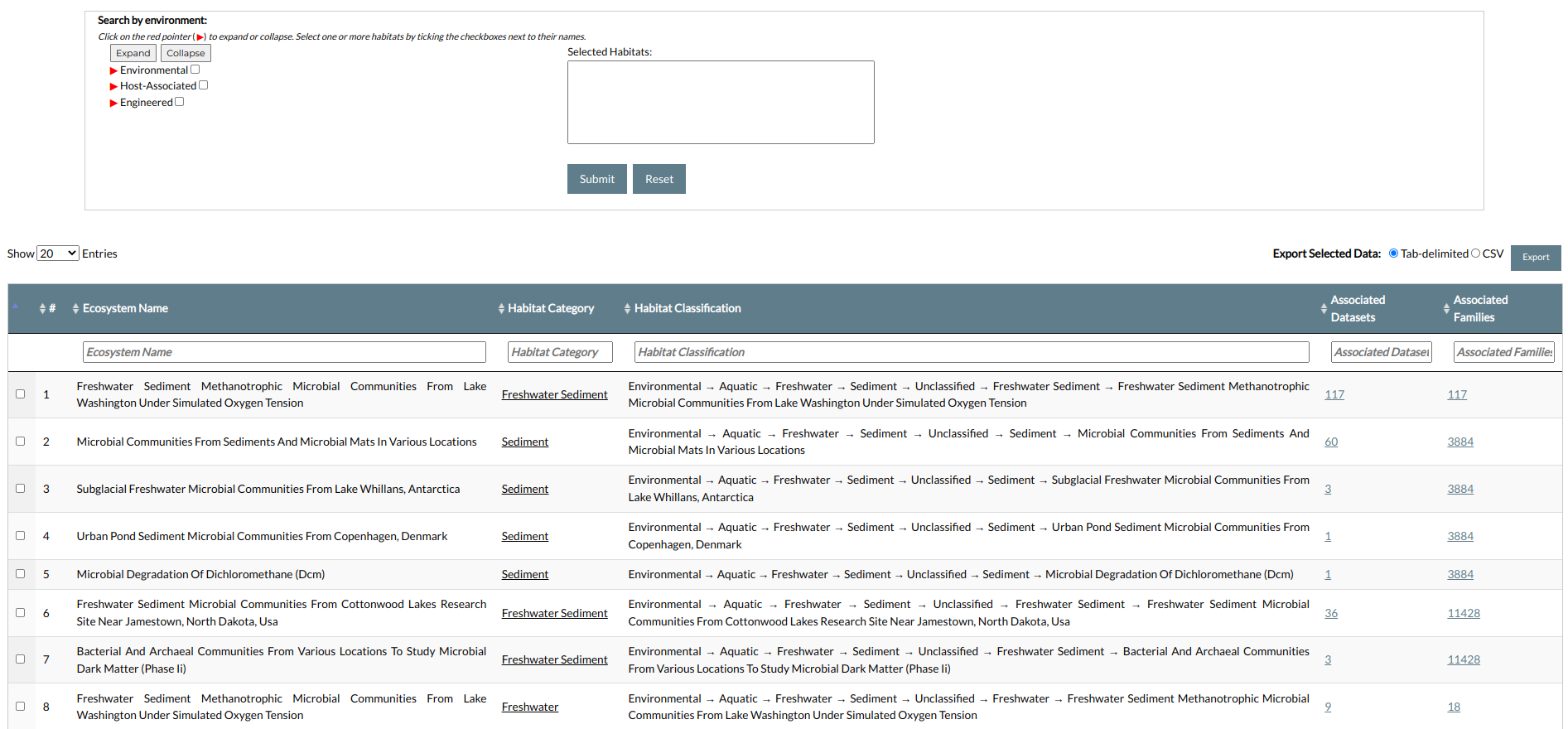
The top of the page contains a search form with a number of options that can help you filter data according to your needs. Browse / search results are presented in a table containing the following information:
- Numeric index: The entry's numeric index in the search
- Ecosystem Name: The name of each unique ecosystem.
- Habitat Type: The type of each ecosystem (e.g. Sediment, Lake, Grass, etc.).
- Habitat Taxonomy: The classification of the ecosystem, from the top level (e.g. Environmental) to the lowest.
- Associated Datasets: the number of associated datasets.
- Associated Families: the number of associated families.
Browse and search results are organized in pages. Users can select the number of entries per page by selecting a value in the “Show Entries” drop-down menu, located at the top left side of the entries table.
One or multiple entries can be selected by clicking their checkboxes, appearing at the start of each line. By clicking the checkbox in the table's head line, all entries per page can be selected at once. Selected entries can then be downloaded, using the options presented at the top right side of the table. Data can be downloaded in tab (TSV) or comma-delimited (CSV) format, by choosing the appropriate option and clicking the “Export” button.
Searches can be performed by using the search form in the top of the page. The form enables you to perform searches using the scaffolds' ecosystem annotation, based on the analyzed metagenome and metatranscriptome datasets. All habitats are shown in a hierarchical tree structure, following the GOLD ecosystem classification system. The available ecosystems belong to one of the following general categories:
- Environmental: contains the ecosystems of datasets coming from environmental samples,e.g. water, soil etc.
- Host-associated: contains ecosystems of datasets obtained from organisms acting as hosts to microbes, e.g. humans, mammals, plants etc.
- Engineered: contains a number of environments that were man-made, e.g. factories, industrial wastes, lab wastes etc.
Each category is further divided into sub-categories that can be displayed or hidden by clicking on the red caret icons. You can also expand or collapse the entire tree by clicking on the Expand All or Collapse All button, respectively.
To select an ecosystem for search, simply click the checkbox next to its name. You can select multiple ecosystems to perform complex queries. The names of the selected ecosystems will appear in the list box at the right of the tree.
Sequence Searches
NMPFamsDB offers a number of tools for performing sequence queries on the database's contents. These can be performed against the NMPF representative consensus sequences, or their Hidden Markov Models (HMMs). The following tools are currently available:
- LAST Search: Perform pairwise sequence alignments in a BLAST-like manner against the NMPF representative sequences.
- HMMER Search: Perform HMM-based operations:
- Sequence vs Sequence: pairwise sequence alignments against the NMPF representative sequences in an HMM-powered iterative manner, with phmmer or jackhmmer.
- Sequence vs HMM: Sequence - HMM queries against the NMPF HMM profiles with hmmscan.
- Pattern Search: search sequence patterns/motifs against the NMPF representative sequences using PROSITE-like patterns or regular expressions.
LAST Search
The LAST search option uses the LAST alignment (lastal) package to perform pairwise sequence alignment searches. LAST operates in a manner very similar to the BLAST and PSI-BLAST algorithms, but has been optimized to work with large datasets and perform searches with a sensitivity matching that of PSI-BLAST.
By choosing Sequence Search → LAST Search you will be redirected to the following input form:

You can perform LAST searches using the following steps:
-
Paste one or more sequences in FASTA format in the text box area, or click the "Choose File" button and upload a FASTA-formatted sequence file. The input can contain one or multiple sequences.
Important Note: These sequences MUST be in the FASTA format: the first line for each sequence contains the header and starts with the ">" character, while the rest of the lines contain the sequence itself, in the one letter aminoacid code. Submitting plain (i.e. no title) sequences WILL NOT WORK.
-
Define the search options: in this step, you need to select a dataset to search against, as well as define the alignment parameters:
2.1 Select Dataset to search: Choose one of the NMPFamsDB available datasets. You can perform searches against the entire set of NMPFs (
All) or choose a subset to limit your searches, e.g.Environmentalfor NMPFs with Environmental ecosystem associations,Metatranscriptome-onlyfor NMPFs containing exclusively metatranscriptome sequences, orBacteriafor NMPFs associated with bacteria.2.2 Substitution Matrix: Select the substitution matrix for the alignments. Available choices include
BLOSUM62(default),BLOSUM80,PAM10,PAM30andMIQS.2.3 Gap Penalties: Select the gap costs for creation (Open) and Extension (Extend). Although you can set the values according to your needs, the default values offered will probably cover most cases. Note that LAST utilizes different default values for different matrix categories, based on their properties:
- BLOSUM62 & BLOSUM80: Open
11, Extend2 - PAM10: Open
20, Extend3 - PAM30: Open
13, Extend3 - MIQS: Open
13, Extend2
- BLOSUM62 & BLOSUM80: Open
-
If you want your results to appear in a new tab or window, click the "Run in New Tab" checkbox.
When you are ready to continue, click the "Submit" button. To clear your choices, click the "Reset" button.
The LAST results are presented in the following window:
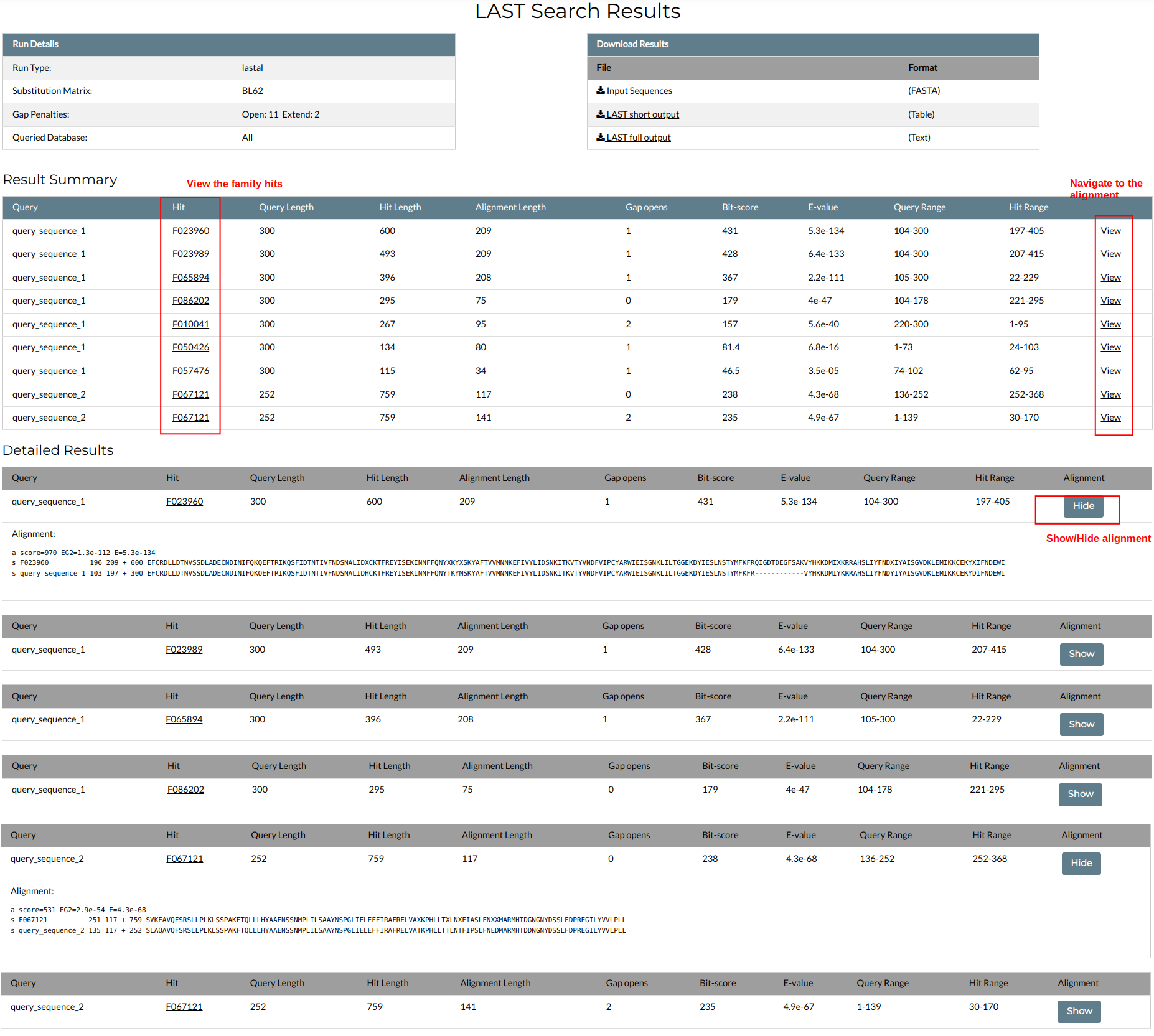
The table at the top left contains the input parameters, i.e. the search dataset, substitution matrix and gap penalties. The table at the top right contains a list with downloadable files, including the input sequence(s) in FASTA format, a short table of results and the full LAST output in text format for parsing.
An overview of the results is given in the Results Summary table. Each hit is represented by a row in the table and includes the sequence name as found in its FASTA header (e.g. query_sequence_1), the ID of the NMPF hit (e.g. F023960), the alignment parameters (query length, hit length, number of gaps, query range and hit range) and scores (Bit-score, E-value, and a link to the alignment itself ("View"). The NMPF IDs are clickable and open the respective NMPFamsDB entry pages. Clicking the "View" link will automatically navigate you to the respective alignment.
Each alingment is presented separately in the Detailed Results section. The title for each alignment contains all the relevant information (Bit-score, E-value etc), while the alignment itself can be shown or hidden by clicking the "Show/Hide" button.
HMMER Search
The HMMER search option uses the HMMER 3 package to perform sequence queries, either with pairwise sequence alignments or with HMM searches.
By choosing Sequence Search → HMMER Search you will be redirected to the following input form:
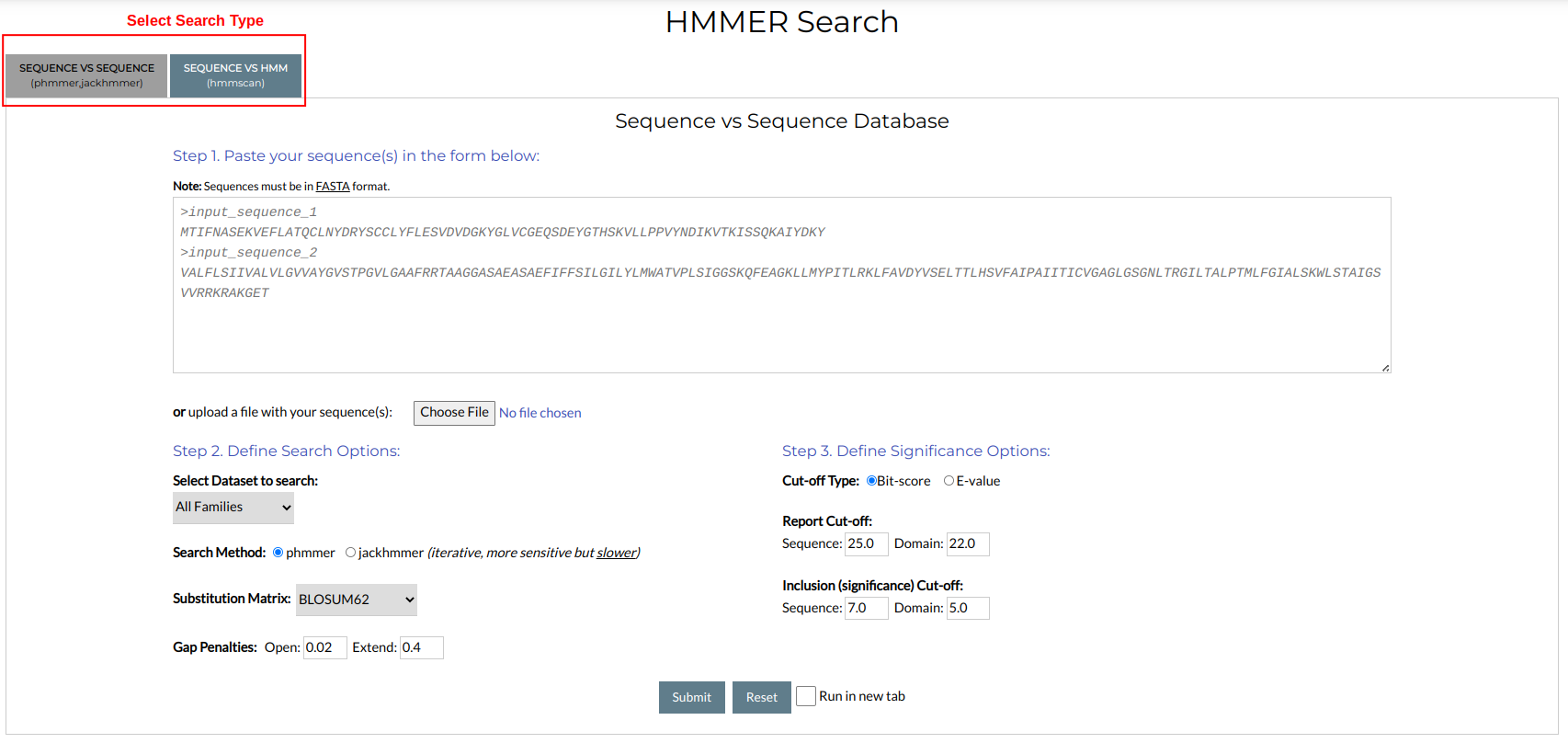
For HMMER, two different search types are offered:
- Sequence vs Sequence: Perform pairwise alignments in an iterative, BLAST-like manner
- Sequence vs HMM: Perform Sequence-HMM searches.
These search types are available by clicking the tab buttons at the top of the form.
Sequence vs Sequence
For Sequence vs Sequence searches perform the following steps:
-
Paste one or more sequences in FASTA format in the text box area, or click the "Choose File" button and upload a FASTA-formatted sequence file. The input can contain one or multiple sequences.
-
Define the search options: in this step, you need to select a dataset to search against, as well as define the alignment parameters:
2.1 Select Dataset to search: Choose one of the NMPFamsDB available datasets. You can perform searches against the entire set of NMPFs (
All) or choose a subset to limit your searches, e.g.Environmentalfor NMPFs with Environmental ecosystem associations,Metatranscriptome-onlyfor NMPFs containing exclusively metatranscriptome sequences, orBacteriafor NMPFs associated with bacteria.2.2 Search Method: Choose the search algorithm you wish to use. Two options are offered,
phmmerandjackhmmer. These are essentially equivalent to the BLAST and PSI-BLAST functions:phmmerperforms pairwise alignments and returns their results, whilejackhmmerfirst performs a pairwise alignment query, uses the results to construct an HMM profile, and then performs a sequence-HMM query using that profile (the same concept as the PSSM matrices used in PSI-BLAST, but this time with HMMs). As expected,phmmeris faster, butjackhmmeris more sensitive to remote homologs.2.3 Substitution Matrix: Select the substitution matrix for the alignments. Available choices include a wide range of BLOSUM and PAM matrices, with
BLOSUM62as the default.2.4 Gap Penalties: Select the gap costs for creation (Open) and Extension (Extend). Although you can set the values according to your needs, the default values offered will probably cover most cases.
-
Define significance options: in this step you define the cut-off values after which results will be deemed as significant and reported. These include the Inclusion and Report thresholds. You can choose whether to base your cut-off on the Bit-score (default) or the E-value. For each case, different default values are used, although you can adjust them to your needs. Note that different thresholds are given for the total sequence alignment (Sequence) and the local domain alignment (Domain); these will also correspond to the reported Bit-scores and E-values for the total sequence and the best domain hits, respectively.
-
If you want your results to appear in a new tab or window, click the "Run in New Tab" checkbox.
When you are ready to continue, click the "Submit" button. To clear your choices, click the "Reset" button.
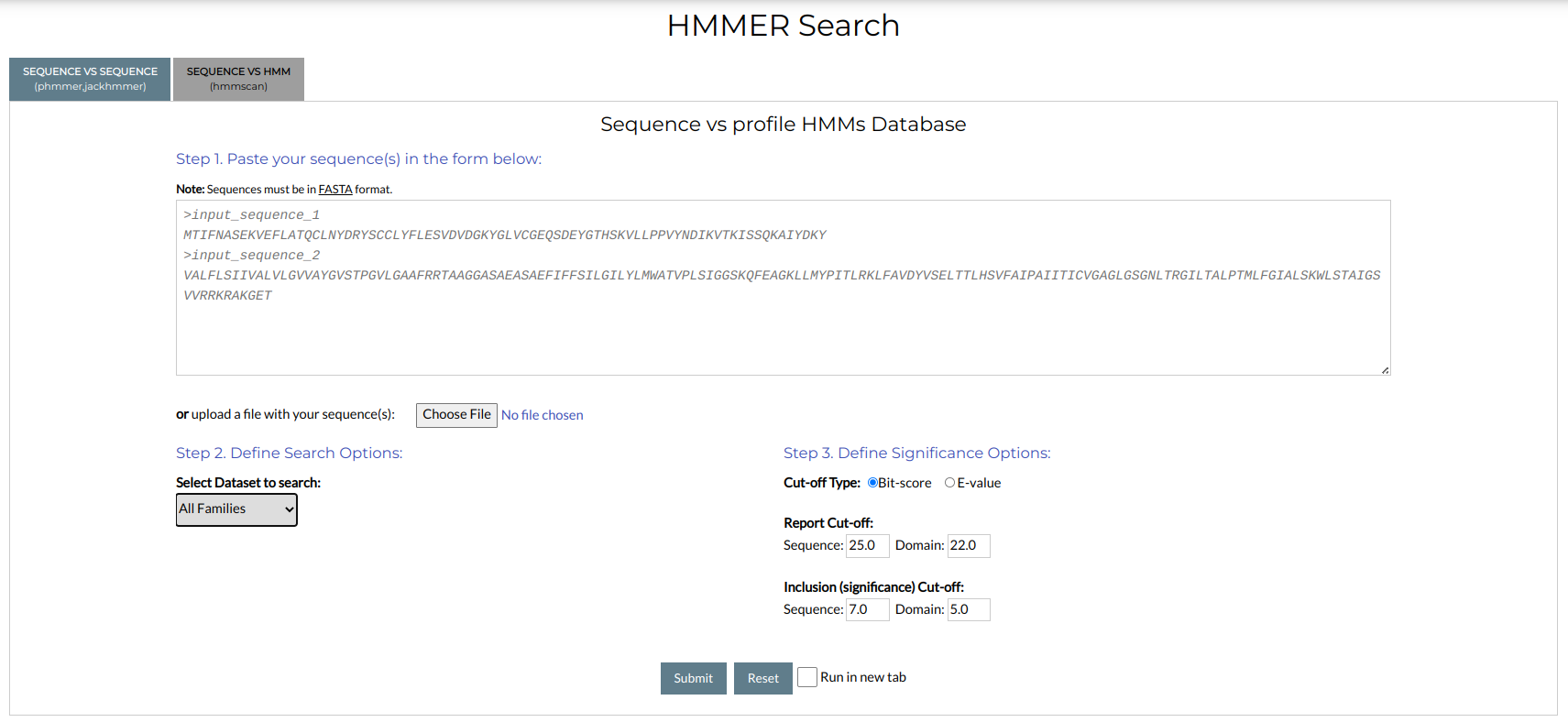
Sequence vs HMM
For Sequence vs HMM searches, select the "Sequence vs HMM" option from the tab menu above the form. You will see the following window:
You can perform a search with the following steps:
-
Paste one or more sequences in FASTA format in the text box area, or click the "Choose File" button and upload a FASTA-formatted sequence file. The input can contain one or multiple sequences.
-
Define the search options: in this step, you need to select a dataset to search against, as well as define the alignment parameters:
2.1 Select Dataset to search: Choose one of the NMPFamsDB available datasets. You can perform searches against the entire set of NMPFs (
All) or choose a subset to limit your searches, e.g.Environmentalfor NMPFs with Environmental ecosystem associations,Metatranscriptome-onlyfor NMPFs containing exclusively metatranscriptome sequences, orBacteriafor NMPFs associated with bacteria. -
Define significance options: in this step you define the cut-off values after which results will be deemed as significant and reported. These include the Inclusion and Report thresholds. You can choose whether to base your cut-off on the Bit-score (default) or the E-value. For each case, different default values are used, although you can adjust them to your needs. Note that different thresholds are given for the total sequence alignment (Sequence) and the local domain alignment (Domain); these will also correspond to the reported Bit-scores and E-values for the total sequence and the best domain hits, respectively.
-
If you want your results to appear in a new tab or window, click the "Run in New Tab" checkbox.
When you are ready to continue, click the "Submit" button. To clear your choices, click the "Reset" button.
The HMMER results, either for Sequence vs Sequence or for Sequence vs HMM search, are presented in a manner similar to LAST:

Ecosystem & Phylogeny
The Ecosystem & Phylogeny tool allows you to visualize and plot the distribution of NMPFs and their relationships, based on their associations with user-defined ecosystems or taxonomic groups. Through the tool, you can create a number of different graph types (matrix plot, venn diagram, circos plot, total-vs-specific bar chart and Upset plot), customize them at will and export them as images.
The tool is available by navigating to Visualization Tools → Ecosystem & Phylogeny. From the top menu. You will be redirected to the following input form:
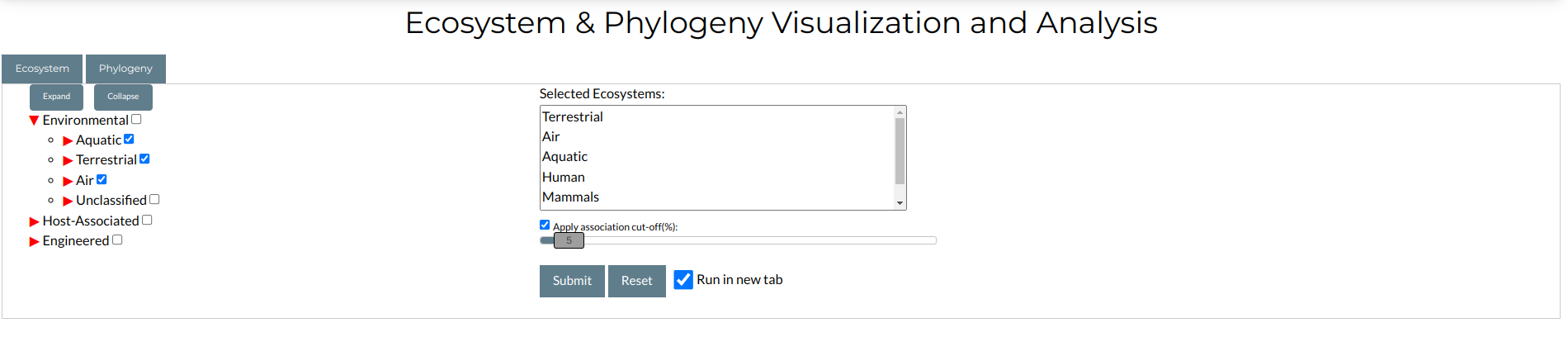
The input has two panels, labeled "Ecosystem" and "Phylogeny" and accessible through the respective tab buttons at the top of the form.
Through "Ecosystem" you can select different ecosystem types, by expanding or collapsing the tree structure at the left of the form and selecting the ecosystems you desire by clicking at the checkbox next to each ecosystem name. The names of the selected ecosystems will appear at the text panel to the right of the form. In addition, you can set an association cut-off, by clicking the Apply association cut-off checkbox and dragging the slider button to the value of your choice (in the above example, we set it at 5 %). Finally, to limit your run to only families with exclusive association to the selected ecosystems (i.e. 100% association cutoff), click on the relevant checkbox.
Similar to "Ecosystem", you can select different taxonomic groups by navigating to the "Phylogeny" tab and using the search form there. The taxonomic groups are organized in a hierarchical tree in the same manner as the ecosystems. An association cutoff can also be set, by clicking the Apply association cut-off checkbox and dragging the slider button to the value of your choice.
When you are ready to begin, click the "Submit" button.
After the tool has finished, you will see the results in the following manner:
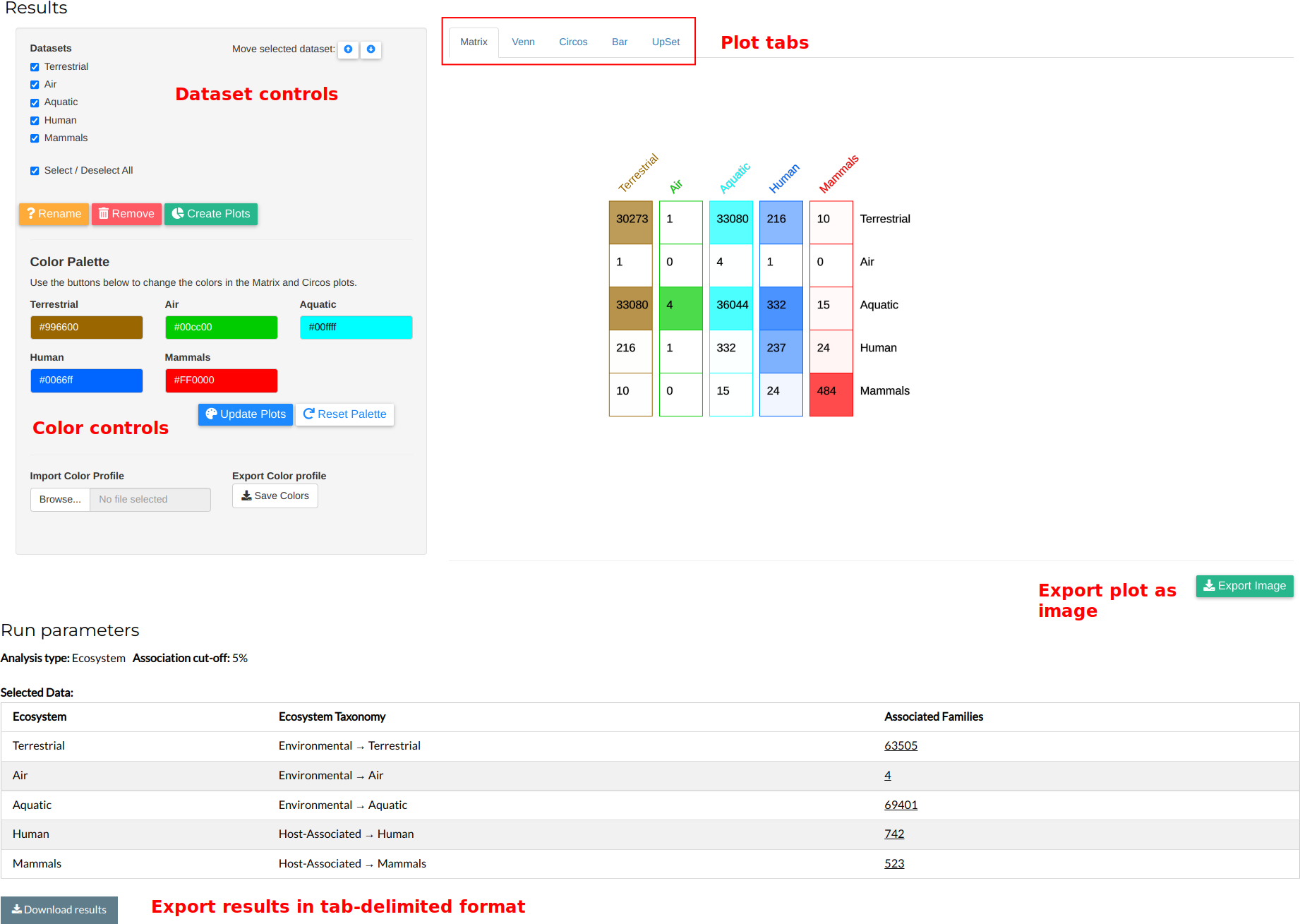
The results are presented in an interactive viewer at the top of the page. These include the run type, association cutoff and a table with all selected categories (ecosystems or taxonomy groups), their taxonomy tree, and number of associated families. The number of families values are links; clicking them will open a new tab into the Browse Families page and retrieve them.
The interactive viewer is vertically split into two parts. To the left, there is a control panel. At the top of the control panel you will find your selected category datasets. You can select or deselect them by clicking on the checkboxes or using the Select / Deselect all checkbox. You can rename or remove them by clicking on the Rename or Remove buttons, respectively. You can change the order of a set in the list by selecting only that set and clicking the Move Up or Move Down buttons at the right of the list. Finally, you can apply your changes by clicking the Create Plots button.
Note: The above actions will re-calculate and re-create all visualizations and plots. This can be time-consuming, especially if the number of families in the datasets is large.
At the bottom of the control panel, you will see a Color Palette panel. Each item in the palette corresponds to a dataset. By clicking on it, a colorpicker window appears, enabling you to change the color. To use the updated colors in your plots, click the Update Plots button. To reset the palette to its original values, click the Reset Palette button. Note that clicking the Update Plots button will only change the color of the plots, not re-calculate them. You can export your color palette to a tab-delimited format using the Export color profile button. You can also import your own color palette by using the Import Color Profile upload button. The palette should be in a tab-delimited format. The first column should contain the dataset names, while the second should contain color values in the hexadecimal format. The first line should contain the column names, Dataset and Color, respectively. An example of a color pallette file is shown below:
Dataset Color
Terrestrial #996600
Air #00CC00
Aquatic #00FFFF
Human #0066FF
Mammals #FF0000The plots of the results are shown to the right of the interactive viewer. Each plot is given in its own subtab, accessible by clicking the tab buttons at the top of the plots panel. At the bottom of each plot, an Export Image button appears, enabling you to download each graph in the PNG image format. The different plot types are shown below:

- A color-coded matrix is a square, N * N map (where N = number of datasets) showing all combinations of family values among the datasets. The names of each set are given in the horizontal and vertical axes, with the number of each cell showing the intersection between two sets (e.g. the intersection between Aquatic and Mammals, in this case, is 15). The diagonal of the matrix presents the dataset-specific components, i.e. the families that appear only in that specific dataset.
- A Venn diagram shows the overlaps between the different sets in a graphical manner. All the different combinations are shown. For reasons of efficiency, Venn diagrams are created for collections of up to 5 datasets.
- The circos plot shows the relationships between the sets in a circular chart.
- The bar plot visualization plots the total vs dataset specific numbers of families in a bar chart.
- The Upset plot is a combinatorial chart, featuring three plots in one: a horizontal bar chart, showing the total components of each dataset, a dot plot, displaying various combinations of sets, and a vertical bar chart, showing the values of each combination.

As the Upset is the most complex of all plot types offered, a number of additional options are given in its panel to allow further customization:
- Type: Change the type of information given. The available options offered are:
- Default: The default type, containing a combination of intersections and unions
- Intersection: show only dataset intersections
- Distinct interesection: show only intersection combinations that appear nowhere else, including the distinct per dataset components
- Distinct per file: Show the distinct per dataset families.
- The above are also connected to the two sliders below:
- Number of datasets in plot: define the number of datasets used in the plot
- Number of datasets in combinations: define the minimum and maximum number of sets in the combinations of the upset.
- Colors: display the chart in color, or in black and white.
- Bar labels: show or hide bar labels.
To update the upset plot click the Update Upset button. Note that this recalculates the values of the upset, so it can be slow, depending on the set sizes.
Geographical Distribution
The Geographical Distribution tool allows you to visualize the distribution of one or more NMPFs on the world map, based on the geolocation metadata of their associated megagenome/metatranscriptome datasets, and the maximum distance among them. The tool operates in the following manner:
- A set of NMPFs (either all families or a selected subset) is defined for investigation, and their associated datasets that have geolocation metadata are retrieved. A maximum distance cut-off (in kilometers) value is set.
- Pairwise distances are calculated among all datasets associated with a family.
- If the family's maximum inter-dataset distance is smaller or equal to the defined cut-off, the family is reported, otherwise, it is discarded.
- The datasets of the reported NMPFs are displayed in an interactive map.
The tool is accessible by navigating to Visualization Tools → Geographical Distribution from the top menu. You will be redirected to the following input form:

The input form has the following two options:
- Select families to analyze: Use the drop-down menu to select if you want to analyze all NMPFs (All) or a selected subset (selected).
- If you choose Selected, an additional form panel will appear to the right, containing the search options for choosing families. These are the same options as in the Browse Families search page and include searching by keywords, sequence & structure features, ecosystem association or phylogeny. You can operate them in the same manner as in searching for families.
- Select maximum dataset distance in family: Use the input field to set a value for the maximum distance between two datasets (in kilometers). The default value is set at 1000 km.
When you are ready, click the Submit button. To clear the input, click Reset. To run the tool in a separate tab, click the Run in new tab check box prior to submitting.
The tool will take some time to finish, during which a loading screen is displayed. When calculations are complete, you will be redirected to the following results page:

The results are divided into two tabs, accessible from the buttons at the top of the panel. The Viewer tab contains the world map, showing the geographical locations of the datasets associated with the families. The map is interactive and can be navigated using the mouse, by left clicking and dragging in the map window. Zooming in and out can be performed by using the mouse scroll button, or by operating the zoom controls at the bottom. In the map, each data point corresponds to a metagenomic dataset; hovering the mouse over it will display a pop-up, showing its associated Taxon OID, longitude and latitude, and the number of associated families. The size of each point is proportional to the number of associated families. Each point is colored based on the classification of its associated ecosystem; with the color index shown at the bottom of the map. A detailed list of the results is given in the Map Points tab. The results are presented in an interactive table, including the IDs of the identified families, the Taxon OIDs of the associated datasets, and their assigned metadata (Longitude, Latitude and ecosystem type).
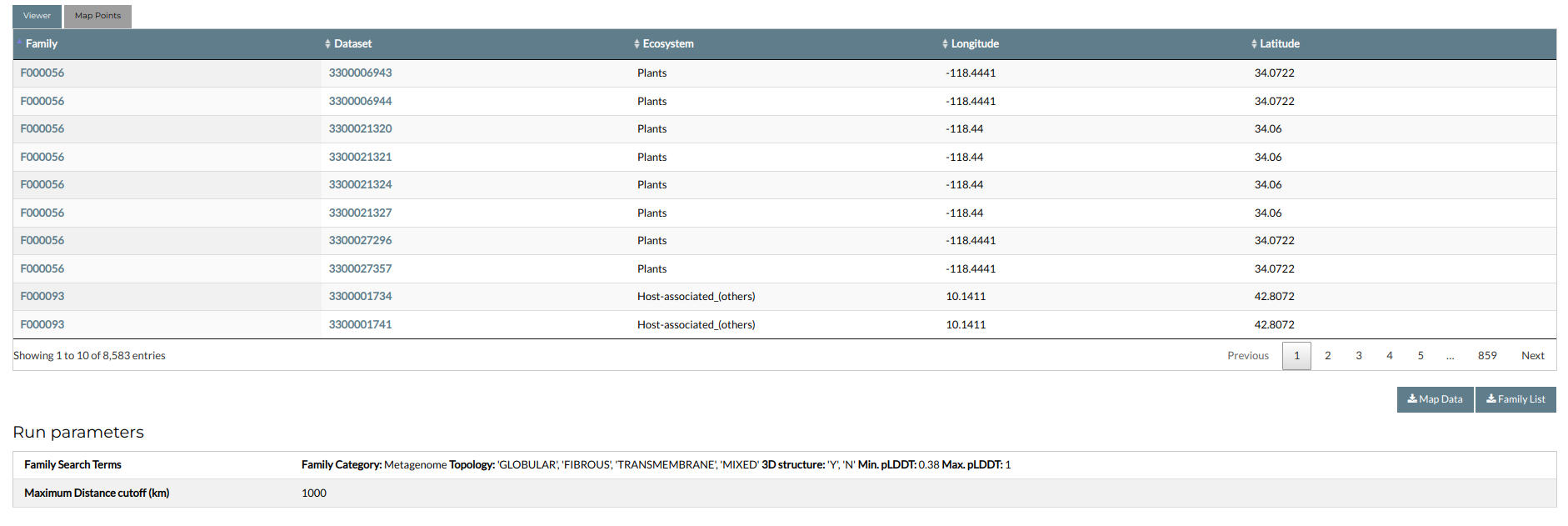
The data can be downloaded by clicking the Map Data button. A list of the families can be downloaded by clicking the Family List button. Finally, the bottom of the page shows the input options, including the family search parameters and the maximum distance cutoff.
Programmatic Access
NMPFamsDB offers two ways of programmatic access to its components:
- Individual Family Entry Data: retrieve entries in various file formats
- Application Programming Interface (API): Access database components and metadata through requests
The first method is easier to use and more immediate, but it is limited to the sequence and structure data of the NMPF families. The second method (the API) is more complex to use; however, it provides access to the full metadata of each NMPF Family entry, as well as the information of the associated metagenome datasets and sequencing scaffolds.
Individual Family Entry Data
In NMPFamsDB, each family entry is represented by a number of files in the following data types:
- Family sequences in the FASTA format
- Full Multiple sequence alignments (MSAs) in the aligned FASTA format
- Non-redundant (seed) MSAS in the aligned FASTA format
- Hidden Markov Models (HMMs) in the HMMER and HH-suite formats
- Sequence logos in the SkyLign JSON format
- 3D structure models in the Protein Data Bank PDB format (Note: these are available only for entries with 3D structure predictions)
Each data type can be accessed through a URL formatted as below:
https://bib.fleming.gr/NMPFamsDB/data/datatype/ID.extension
where:
-
datatypeis the type of the data file:- Sequences:
fasta - Full MSAs:
aln - Seed MSAs:
seed - HMMER HMMs:
hmm - HH-suite HMMs:
hhm - Sequence Logos:
logo - AlphaFold2 3D models:
pdb - Processed "Pivot" MSAs used as AlphaFold2 input:
pivot
- Sequences:
-
IDis the NMPFamsDB family identifier -
extensionis the file extension:- Sequences and MSAs:
fasta - HMMER:
hmm - HH-suite:
hhm - Sequence Logos:
json - 3D models:
pdb
- Sequences and MSAs:
For example, to automatically retrieve the components of entry F000872, you can use the following links:
https://bib.fleming.gr/NMPFamsDB/data/fasta/F000872.fasta https://bib.fleming.gr/NMPFamsDB/data/aln/F000872.fasta https://bib.fleming.gr/NMPFamsDB/data/seed/F000872.fasta https://bib.fleming.gr/NMPFamsDB/data/hmm/F000872.hmm https://bib.fleming.gr/NMPFamsDB/data/hhm/F000872.hhm https://bib.fleming.gr/NMPFamsDB/data/logo/F000872.json https://bib.fleming.gr/NMPFamsDB/data/pdb/F000872.pdb https://bib.fleming.gr/NMPFamsDB/data/pivot/F000872.fasta
The above can be typed as addresses in a browser, or automatically downloaded with utilities such as wget or cURL.
Application Programming Interface (API)
NMPFamsDB offers an Application Programming Interface (API), enabling users to retrieve database components without utilizing the web interface. Through the API, you can programmatically access subsets of information and retrieve their components, access the database with scripts written in various languages like Perl, Python, R etc., or incorporate connections with NMPFamsDB to your own applications or web pages.
The API currently serves results in the JSON format. All API services can be accessed by using both GET and POST requests.
A detailed description can be found in the dedicated API reference page.
Downloads
The Downloads page contains all NMPF information for download in the following formats:
-
Sequences: The novel protein sequences in the FASTA format. The sequences of each NMPF are given in a distinct FASTA file (e.g.
F000001.fasta). -
Full MSAs: The full MSAs of the families in the FASTA format. Each MSA is represented by a distinct file (e.g.
F000001_msa.fasta). -
Seed MSAs: The non-redundant, seed MSAs of the families in the FASTA format. Each MSA is represented by a distinct file (e.g.
F000001_seed.fasta). -
Consensus Sequences: The consesus sequences of the NMPFs, in a signle FASTA file (
consensus.fasta). -
HMM profiles: The Hidden Markov Models in the HMMER format. Each NMPF is contained in a distinct file (e.g.
F105879.hmm). -
3D Models: The NMPFs with available 3D structures in the PDB format.
The download options are given for the entire NMPFamsDB contents (All), as well as its following subsets:
-
Clustered based on habitat distribution:
-
Environmental: A collection of NMPFs that are associated with Environmental ecosystems.
-
Host-associated: A collection of NMPFs that are associated with Host-associated ecosystems.
-
Engineered: A collection of NMPFs that are associated with Engineered ecosystems.
-
-
Clustered based on phylogeny:
-
Bacteria: A collection of NMPFs that are associated with Bacteria.
-
Archaea: A collection of NMPFs that are associated with Archaea.
-
Eukaryota: A collection of NMPFs that are associated with Eukaryota.
-
Viruses: A collection of NMPFs that are associated with viruses.
-
Unclassified: A collection of NMPFs that have unclassified sequences.
-
-
Clustered based on sample type:
-
Metagenome: Metagenome-only NMPFs
-
Metatranscriptome: Metatranscriptome-only NMPFs
-
Metagenome/Metatranscriptome: Mixed Metagenome/Metatranscriptome NMPFs
-
All of the aforementioned downloads are given in compressed (gzip) tar archives. They can be opened with graphical applications such as WinRar, 7-zip, Engrampa etc., or with the use of the tar command line (tar xvf package.tar.gz).
In addition, the original sequence data, as retrieved from IMG/M and clustered, are given for download in tab-delimited format. For each sequence, the header, containing the Taxon OID, Scaffold ID, Gene ID and Family, separated by pipes ("|"), are given in the first column, and the sequence in the second. Tab-delimited downloads are given for the following two datasets:
-
Metagenome Families: The metagenomic sequences that were clustered into NMPFs
-
Reference Genome Families: A sequence dataset derived from IMG's reference genomes, and clustered in the same manner as the metagenomes, for comparison. See Pavlopoulos et. al, 2022 for more details.
Privacy Policy
The administrators of NMPFamDB, in accordance with Regulation (EU) 2016/679 and the relevant national legislation on the protection of natural persons with regard to the processing of personal data, provide the following privacy notice to explain what personal data is collected, for what purposes, how it is processed and how we keep it secure.
1. Who controls your personal data and how to contact us?
All data are collected and processed by the administrators of the NMPFamDB database. You can contact us using the information provided in the Contact page.
2. What is the lawful basis for data collection?
Data are collected to help monitor website functionality, resolve issues, improve the allocated resources and provide services to you adequately.
3. What personal data is collected from users?
The personal data collected by the website’s services are as follows:
- IP address
- Date and time of a visit to the service
- Operating System
- Browser
- Amount of data transmitted
The data administrators use the aforementioned data for the following processes:
- To provide the user access to the service
- To conduct and monitor data protection activities
- To conduct and monitor website security
- To better understand the needs of the users and guide future improvements of the service
- To communicate with users and answer their questions
4. Who has access to your personal data?
Any collected personal data is solely accessed and controlled by the website’s administrators (see question 1). No other person has access to the data.
5. Will your personal data be transferred to other organisations?
Any personal data directly collected by NMPFamDB's services are handled by the administrators of NMPFamDB exclusively. There are no transfers to any other organisations whatsoever for these data.
Please note that NMPFamDB utilizes a number of third-party resources to provide you with the best possible experience. These include jQuery, FontAwesome, Google Fonts and OpenStreetMap/OpenLayers. Some of these resources store cookies and may record some data to function. The administrators of NMPFamDB are not responsible for the treatment of any data by these services. You are advised to consult the Privacy Policies of each of these services through their respective web pages.
6. How long is your personal data kept?
Any personal data directly obtained from you will be retained for the minimum amount of time possible to ensure legal compliance and to facilitate internal and external audits if they arise.
7. Cookies Policy
NMPFamDB uses cookies to achieve functionality and provide you with the best possible experience. Specifically, we use cookies for the following purposes:
- Functionality: we use cookies for the proper functionality of the web services offered by NMPFamDB.
- Security: we use cookies as part of the security measures aimedat protecting your privacy, and our website and services generally.
- Cookie consent: we use cookies to store your preferences in relation to the use of cookies more generally.
Most browsers allow you to refuse to accept cookies and to delete cookies. The methods for doing so vary from browser to browser, and from version to version. You can however obtain up-to-date information about blocking and deleting cookies via these links:
Blocking all cookies will have a negative impact upon the usability of NMPFamDB.
8. The website’s data controllers provide the following rights regarding your personal data
You have the right to:
- Not be subject to decisions based solely on an automated processing of data (i.e. without human intervention) without you having your views taken into consideration.
- Request at reasonable intervals and without excessive delay or expense, information about the personal data processed about you. Under your request we will inform you in writing about, for example, the origin of the personal data or the preservation period.
- Request information to understand data processing activities when the results of these activities are applied to you.
- Object at any time to the processing of your personal data unless we can demonstrate that we have legitimate reasons to process your personal data.
- Request free of charge and without excessive delay rectification or erasure of your personal data if we have not been processing it respecting the data protection policies of the respective controllers.
It must be clarified that rights 4 and 5 are only available whenever the processing of your personal data is not necessary to:
- Comply with a legal obligation.
- Perform a task carried out in the public interest.
- Exercise authority as a data controller.
- Archive for purposes in the public interest, or for historical research purposes, or for statistical purposes.
- Establish, exercise or defend legal claims.
Any requests and objections can be sent to us through the Contact page.
